
Justin Crocker's reading list...
Saturday, March 30, 2013
Thursday, March 28, 2013
Precise and reliable gene expression via standard transcription and translation initiation elements
Precise and reliable gene expression via standard transcription and translation initiation elements:
Nature Methods 10, 354 (2013).
doi:10.1038/nmeth.2404
Authors: Vivek K Mutalik, Joao C Guimaraes, Guillaume Cambray, Colin Lam, Marc Juul Christoffersen, Quynh-Anh Mai, Andrew B Tran, Morgan Paull, Jay D Keasling, Adam P Arkin & Drew Endy

Nature Methods 10, 354 (2013).
doi:10.1038/nmeth.2404
Authors: Vivek K Mutalik, Joao C Guimaraes, Guillaume Cambray, Colin Lam, Marc Juul Christoffersen, Quynh-Anh Mai, Andrew B Tran, Morgan Paull, Jay D Keasling, Adam P Arkin & Drew Endy
Tuesday, March 26, 2013
Dpp induction of vn initiates wing development [Developmental Biology]
Dpp induction of vn initiates wing development [Developmental Biology]: The acquisition of flight contributed to the success of insects and winged forms are present in most orders. Key to understanding the origin of wings will be knowledge of the earliest postembryonic events promoting wing outgrowth. The Drosophila melanogaster wing is intensely studied as a model appendage, and yet little...

A Conserved Role for Human Nup98 in Altering Chromatin Structure and Promoting Epigenetic Transcriptional Memory
A Conserved Role for Human Nup98 in Altering Chromatin Structure and Promoting Epigenetic Transcriptional Memory:
by William H. Light, Jonathan Freaney, Varun Sood, Abbey Thompson, Agustina D'Urso, Curt M. Horvath, Jason H. Brickner

The interaction of nuclear pore proteins (Nups) with active genes can promote their transcription. In yeast, some inducible genes interact with the nuclear pore complex both when active and for several generations after being repressed, a phenomenon called epigenetic transcriptional memory. This interaction promotes future reactivation and requires Nup100, a homologue of human Nup98. A similar phenomenon occurs in human cells; for at least four generations after treatment with interferon gamma (IFN-γ), many IFN-γ-inducible genes are induced more rapidly and more strongly than in cells that have not previously been exposed to IFN-γ. In both yeast and human cells, the recently expressed promoters of genes with memory exhibit persistent dimethylation of histone H3 lysine 4 (H3K4me2) and physically interact with Nups and a poised form of RNA polymerase II. However, in human cells, unlike yeast, these interactions occur in the nucleoplasm. In human cells transiently depleted of Nup98 or yeast cells lacking Nup100, transcriptional memory is lost; RNA polymerase II does not remain associated with promoters, H3K4me2 is lost, and the rate of transcriptional reactivation is reduced. These results suggest that Nup100/Nup98 binding to recently expressed promoters plays a conserved role in promoting epigenetic transcriptional memory.
by William H. Light, Jonathan Freaney, Varun Sood, Abbey Thompson, Agustina D'Urso, Curt M. Horvath, Jason H. Brickner
The interaction of nuclear pore proteins (Nups) with active genes can promote their transcription. In yeast, some inducible genes interact with the nuclear pore complex both when active and for several generations after being repressed, a phenomenon called epigenetic transcriptional memory. This interaction promotes future reactivation and requires Nup100, a homologue of human Nup98. A similar phenomenon occurs in human cells; for at least four generations after treatment with interferon gamma (IFN-γ), many IFN-γ-inducible genes are induced more rapidly and more strongly than in cells that have not previously been exposed to IFN-γ. In both yeast and human cells, the recently expressed promoters of genes with memory exhibit persistent dimethylation of histone H3 lysine 4 (H3K4me2) and physically interact with Nups and a poised form of RNA polymerase II. However, in human cells, unlike yeast, these interactions occur in the nucleoplasm. In human cells transiently depleted of Nup98 or yeast cells lacking Nup100, transcriptional memory is lost; RNA polymerase II does not remain associated with promoters, H3K4me2 is lost, and the rate of transcriptional reactivation is reduced. These results suggest that Nup100/Nup98 binding to recently expressed promoters plays a conserved role in promoting epigenetic transcriptional memory.
Friday, March 22, 2013
Divergent Selection Drives Genetic Differentiation in an R2R3-MYB Transcription Factor That Contributes to Incipient Speciation in Mimulus aurantiacus
Divergent Selection Drives Genetic Differentiation in an R2R3-MYB Transcription Factor That Contributes to Incipient Speciation in Mimulus aurantiacus:
by Matthew A. Streisfeld, Wambui N. Young, James M. Sobel

Identifying the molecular genetic basis of traits contributing to speciation is of crucial importance for understanding the ecological and evolutionary mechanisms that generate biodiversity. Despite several examples describing putative “speciation genes,” it is often uncertain to what extent these genetic changes have contributed to gene flow reductions in nature. Therefore, considerable interest lies in characterizing the molecular basis of traits that actively confer reproductive isolation during the early stages of speciation, as these loci can be attributed directly to the process of divergence. In Southern California, two ecotypes of Mimulus aurantiacus are parapatric and differ primarily in flower color, with an anthocyanic, red-flowered morph in the west and an anthocyanin-lacking, yellow-flowered morph in the east. Evidence suggests that the genetic changes responsible for this shift in flower color have been essential for divergence and have become fixed in natural populations of each ecotype due to almost complete differences in pollinator preference. In this study, we demonstrate that a cis-regulatory mutation in an R2R3-MYB transcription factor results in differential regulation of enzymes in the anthocyanin biosynthetic pathway and is the major contributor to differences in floral pigmentation. In addition, molecular population genetic data show that, despite gene flow at neutral loci, divergent selection has driven the fixation of alternate alleles at this gene between ecotypes. Therefore, by identifying the genetic basis underlying ecologically based divergent selection in flower color between these ecotypes, we have revealed the ecological and functional mechanisms involved in the evolution of pre-mating isolation at the early stages of incipient speciation.
by Matthew A. Streisfeld, Wambui N. Young, James M. Sobel
Identifying the molecular genetic basis of traits contributing to speciation is of crucial importance for understanding the ecological and evolutionary mechanisms that generate biodiversity. Despite several examples describing putative “speciation genes,” it is often uncertain to what extent these genetic changes have contributed to gene flow reductions in nature. Therefore, considerable interest lies in characterizing the molecular basis of traits that actively confer reproductive isolation during the early stages of speciation, as these loci can be attributed directly to the process of divergence. In Southern California, two ecotypes of Mimulus aurantiacus are parapatric and differ primarily in flower color, with an anthocyanic, red-flowered morph in the west and an anthocyanin-lacking, yellow-flowered morph in the east. Evidence suggests that the genetic changes responsible for this shift in flower color have been essential for divergence and have become fixed in natural populations of each ecotype due to almost complete differences in pollinator preference. In this study, we demonstrate that a cis-regulatory mutation in an R2R3-MYB transcription factor results in differential regulation of enzymes in the anthocyanin biosynthetic pathway and is the major contributor to differences in floral pigmentation. In addition, molecular population genetic data show that, despite gene flow at neutral loci, divergent selection has driven the fixation of alternate alleles at this gene between ecotypes. Therefore, by identifying the genetic basis underlying ecologically based divergent selection in flower color between these ecotypes, we have revealed the ecological and functional mechanisms involved in the evolution of pre-mating isolation at the early stages of incipient speciation.
Genome-Wide Control of RNA Polymerase II Activity by Cohesin
Genome-Wide Control of RNA Polymerase II Activity by Cohesin:
by Cheri A. Schaaf, Hojoong Kwak, Amanda Koenig, Ziva Misulovin, David W. Gohara, Audrey Watson, Yanjiao Zhou, John T. Lis, Dale Dorsett

Cohesin is a well-known mediator of sister chromatid cohesion, but it also influences gene expression and development. These non-canonical roles of cohesin are not well understood, but are vital: gene expression and development are altered by modest changes in cohesin function that do not disrupt chromatid cohesion. To clarify cohesin's roles in transcription, we measured how cohesin controls RNA polymerase II (Pol II) activity by genome-wide chromatin immunoprecipitation and precision global run-on sequencing. On average, cohesin-binding genes have more transcriptionally active Pol II and promoter-proximal Pol II pausing than non-binding genes, and are more efficient, producing higher steady state levels of mRNA per transcribing Pol II complex. Cohesin depletion frequently decreases gene body transcription but increases pausing at cohesin-binding genes, indicating that cohesin often facilitates transition of paused Pol II to elongation. In many cases, this likely reflects a role for cohesin in transcriptional enhancer function. Strikingly, more than 95% of predicted extragenic enhancers bind cohesin, and cohesin depletion can reduce their association with Pol II, indicating that cohesin facilitates enhancer-promoter contact. Cohesin depletion decreases the levels of transcriptionally engaged Pol II at the promoters of most genes that don't bind cohesin, suggesting that cohesin controls expression of one or more broadly acting general transcription factors. The multiple transcriptional roles of cohesin revealed by these studies likely underlie the growth and developmental deficits caused by minor changes in cohesin activity.
by Cheri A. Schaaf, Hojoong Kwak, Amanda Koenig, Ziva Misulovin, David W. Gohara, Audrey Watson, Yanjiao Zhou, John T. Lis, Dale Dorsett
Cohesin is a well-known mediator of sister chromatid cohesion, but it also influences gene expression and development. These non-canonical roles of cohesin are not well understood, but are vital: gene expression and development are altered by modest changes in cohesin function that do not disrupt chromatid cohesion. To clarify cohesin's roles in transcription, we measured how cohesin controls RNA polymerase II (Pol II) activity by genome-wide chromatin immunoprecipitation and precision global run-on sequencing. On average, cohesin-binding genes have more transcriptionally active Pol II and promoter-proximal Pol II pausing than non-binding genes, and are more efficient, producing higher steady state levels of mRNA per transcribing Pol II complex. Cohesin depletion frequently decreases gene body transcription but increases pausing at cohesin-binding genes, indicating that cohesin often facilitates transition of paused Pol II to elongation. In many cases, this likely reflects a role for cohesin in transcriptional enhancer function. Strikingly, more than 95% of predicted extragenic enhancers bind cohesin, and cohesin depletion can reduce their association with Pol II, indicating that cohesin facilitates enhancer-promoter contact. Cohesin depletion decreases the levels of transcriptionally engaged Pol II at the promoters of most genes that don't bind cohesin, suggesting that cohesin controls expression of one or more broadly acting general transcription factors. The multiple transcriptional roles of cohesin revealed by these studies likely underlie the growth and developmental deficits caused by minor changes in cohesin activity.
Single-Cell Dynamics of Genome-Nuclear Lamina Interactions
Single-Cell Dynamics of Genome-Nuclear Lamina Interactions: Jop Kind, Ludo Pagie, Havva Ortabozkoyun, Shelagh Boyle, Sandra S. de Vries, Hans Janssen, Mario Amendola, Leisha D. Nolen, Wendy A. Bickmore, Bas van Steensel. The nuclear lamina (NL) interacts with hundreds of large genomic regions termed lamina associated domains (LADs). The dynamics of these interactions and the relation to epigenetic modifications ar....
Thursday, March 21, 2013
[Report] Emergence and Diversification of Fly Pigmentation Through Evolution of a Gene Regulatory Module
[Report] Emergence and Diversification of Fly Pigmentation Through Evolution of a Gene Regulatory Module: Pigmentation spots on the wings of flies originate from changes at different levels of the underlying genetic hierarchy.
Authors: Laurent Arnoult, Kathy F. Y. Su, Diogo Manoel, Caroline Minervino, Justine Magriña, Nicolas Gompel, Benjamin Prud’homme
Authors: Laurent Arnoult, Kathy F. Y. Su, Diogo Manoel, Caroline Minervino, Justine Magriña, Nicolas Gompel, Benjamin Prud’homme
Wednesday, March 20, 2013
Circular RNAs are a large class of animal RNAs with regulatory potency
Circular RNAs are a large class of animal RNAs with regulatory potency:
Circular RNAs are a large class of animal RNAs with regulatory potency
Nature 495, 7441 (2013). doi:10.1038/nature11928
Authors: Sebastian Memczak, Marvin Jens, Antigoni Elefsinioti, Francesca Torti, Janna Krueger, Agnieszka Rybak, Luisa Maier, Sebastian D. Mackowiak, Lea H. Gregersen, Mathias Munschauer, Alexander Loewer, Ulrike Ziebold, Markus Landthaler, Christine Kocks, Ferdinand le Noble & Nikolaus Rajewsky
Circular RNAs (circRNAs) in animals are an enigmatic class of RNA with unknown function. To explore circRNAs systematically, we sequenced and computationally analysed human, mouse and nematode RNA. We detected thousands of well-expressed, stable circRNAs, often showing tissue/developmental-stage-specific expression. Sequence analysis indicated important regulatory functions

Circular RNAs are a large class of animal RNAs with regulatory potency
Nature 495, 7441 (2013). doi:10.1038/nature11928
Authors: Sebastian Memczak, Marvin Jens, Antigoni Elefsinioti, Francesca Torti, Janna Krueger, Agnieszka Rybak, Luisa Maier, Sebastian D. Mackowiak, Lea H. Gregersen, Mathias Munschauer, Alexander Loewer, Ulrike Ziebold, Markus Landthaler, Christine Kocks, Ferdinand le Noble & Nikolaus Rajewsky
Circular RNAs (circRNAs) in animals are an enigmatic class of RNA with unknown function. To explore circRNAs systematically, we sequenced and computationally analysed human, mouse and nematode RNA. We detected thousands of well-expressed, stable circRNAs, often showing tissue/developmental-stage-specific expression. Sequence analysis indicated important regulatory functions
Monday, March 18, 2013
Enhancers: five essential questions
Enhancers: five essential questions:
Nature Reviews Genetics 14, 288 (2013).
doi:10.1038/nrg3458
Authors: Len A. Pennacchio, Wendy Bickmore, Ann Dean, Marcelo A. Nobrega & Gill Bejerano
It is estimated that the human genome contains hundreds of thousands of enhancers, so understanding these gene-regulatory elements is a crucial goal. Several fundamental questions need to be addressed about enhancers, such as how do we identify them all, how do they work, and how do they contribute to disease and evolution? Five prominent researchers in this field look at how much we know already and what needs to be done to answer these questions.

Nature Reviews Genetics 14, 288 (2013).
doi:10.1038/nrg3458
Authors: Len A. Pennacchio, Wendy Bickmore, Ann Dean, Marcelo A. Nobrega & Gill Bejerano
It is estimated that the human genome contains hundreds of thousands of enhancers, so understanding these gene-regulatory elements is a crucial goal. Several fundamental questions need to be addressed about enhancers, such as how do we identify them all, how do they work, and how do they contribute to disease and evolution? Five prominent researchers in this field look at how much we know already and what needs to be done to answer these questions.
Gene expression: Influences on noise
Gene expression: Influences on noise:
Nature Reviews Genetics 14, 238 (2013).
doi:10.1038/nrg3448
Author: Mary Muers
Studies of transcription at a single-cell level have revealed extensive cell-to-cell variability in gene expression levels: a phenomenon known as transcriptional noise. Gene expression is influenced by trans-acting factors and cis-acting regulatory elements, and it is known that some sequence elements in promoters

Nature Reviews Genetics 14, 238 (2013).
doi:10.1038/nrg3448
Author: Mary Muers
Studies of transcription at a single-cell level have revealed extensive cell-to-cell variability in gene expression levels: a phenomenon known as transcriptional noise. Gene expression is influenced by trans-acting factors and cis-acting regulatory elements, and it is known that some sequence elements in promoters
Friday, March 15, 2013
Strong Mutational Bias Toward Deletions in the Drosophila melanogaster Genome Is Compensated by Selection
Strong Mutational Bias Toward Deletions in the Drosophila melanogaster Genome Is Compensated by Selection:
Insertions and deletions (collectively indels) obviously have a major impact on genome evolution. However, before large-scale data on indel polymorphism became available, it was difficult to estimate the strength of selection acting on indel mutations. Here, we analyze indel polymorphism and divergence in different compartments of the Drosophila melanogaster genome: exons, introns of different lengths, and intergenic regions. Data on low-frequency polymorphisms indicate that 0.036–0.039 short (1–30 nt) insertion mutations and 0.085–0.092 short deletion mutations, with mean lengths 3.23 and 4.78, respectively, occur per single-nucleotide substitution. The excess of short deletion over short insertion mutations implies that indel mutations of these lengths should lead to a loss of approximately 0.30 nt per single-nucleotide replacement. However, polymorphism and divergence data show that this deletion bias is almost completely compensated by selection: Negative selection is stronger against deletions, whereas insertions are more likely to be favored by positive selection. Among the inframe low-frequency polymorphic mutations in exons, long introns, and intergenic regions, selection prevents a larger fraction of deletions (80–87%, depending on the type of the compartment) than of insertions (70–82%) or single-nucleotide substitutions (49–73%), from reaching high frequencies. The corresponding fractions were the lowest in short introns: 66%, 47%, and 15%, respectively, consistent with the weakest selective constraint in them. The McDonald–Kreitman test shows that 32–46% of the deletions and 60–73% of the insertions that were fixed in the recent evolution of D. melanogaster are adaptive, whereas this fraction is only 0–29% for single-nucleotide substitutions.
Insertions and deletions (collectively indels) obviously have a major impact on genome evolution. However, before large-scale data on indel polymorphism became available, it was difficult to estimate the strength of selection acting on indel mutations. Here, we analyze indel polymorphism and divergence in different compartments of the Drosophila melanogaster genome: exons, introns of different lengths, and intergenic regions. Data on low-frequency polymorphisms indicate that 0.036–0.039 short (1–30 nt) insertion mutations and 0.085–0.092 short deletion mutations, with mean lengths 3.23 and 4.78, respectively, occur per single-nucleotide substitution. The excess of short deletion over short insertion mutations implies that indel mutations of these lengths should lead to a loss of approximately 0.30 nt per single-nucleotide replacement. However, polymorphism and divergence data show that this deletion bias is almost completely compensated by selection: Negative selection is stronger against deletions, whereas insertions are more likely to be favored by positive selection. Among the inframe low-frequency polymorphic mutations in exons, long introns, and intergenic regions, selection prevents a larger fraction of deletions (80–87%, depending on the type of the compartment) than of insertions (70–82%) or single-nucleotide substitutions (49–73%), from reaching high frequencies. The corresponding fractions were the lowest in short introns: 66%, 47%, and 15%, respectively, consistent with the weakest selective constraint in them. The McDonald–Kreitman test shows that 32–46% of the deletions and 60–73% of the insertions that were fixed in the recent evolution of D. melanogaster are adaptive, whereas this fraction is only 0–29% for single-nucleotide substitutions.
Thursday, March 14, 2013
Microhomology-Mediated Mechanisms Underlie Non-Recurrent Disease-Causing Microdeletions of the FOXL2 Gene or Its Regulatory Domain
Microhomology-Mediated Mechanisms Underlie Non-Recurrent Disease-Causing Microdeletions of the FOXL2 Gene or Its Regulatory Domain:
by Hannah Verdin, Barbara D'haene, Diane Beysen, Yana Novikova, Björn Menten, Tom Sante, Pablo Lapunzina, Julian Nevado, Claudia M. B. Carvalho, James R. Lupski, Elfride De Baere
Genomic disorders are often caused by recurrent copy number variations (CNVs), with nonallelic homologous recombination (NAHR) as the underlying mechanism. Recently, several microhomology-mediated repair mechanisms—such as microhomology-mediated end-joining (MMEJ), fork stalling and template switching (FoSTeS), microhomology-mediated break-induced replication (MMBIR), serial replication slippage (SRS), and break-induced SRS (BISRS)—were described in the etiology of non-recurrent CNVs in human disease. In addition, their formation may be stimulated by genomic architectural features. It is, however, largely unexplored to what extent these mechanisms contribute to rare, locus-specific pathogenic CNVs. Here, fine-mapping of 42 microdeletions of the FOXL2 locus, encompassing FOXL2 (32) or its regulatory domain (10), serves as a model for rare, locus-specific CNVs implicated in genetic disease. These deletions lead to blepharophimosis syndrome (BPES), a developmental condition affecting the eyelids and the ovary. For breakpoint mapping we used targeted array-based comparative genomic hybridization (aCGH), quantitative PCR (qPCR), long-range PCR, and Sanger sequencing of the junction products. Microhomology, ranging from 1 bp to 66 bp, was found in 91.7% of 24 characterized breakpoint junctions, being significantly enriched in comparison with a random control sample. Our results show that microhomology-mediated repair mechanisms underlie at least 50% of these microdeletions. Moreover, genomic architectural features, like sequence motifs, non-B DNA conformations, and repetitive elements, were found in all breakpoint regions. In conclusion, the majority of these microdeletions result from microhomology-mediated mechanisms like MMEJ, FoSTeS, MMBIR, SRS, or BISRS. Moreover, we hypothesize that the genomic architecture might drive their formation by increasing the susceptibility for DNA breakage or promote replication fork stalling. Finally, our locus-centered study, elucidating the etiology of a large set of rare microdeletions involved in a monogenic disorder, can serve as a model for other clustered, non-recurrent microdeletions in genetic disease.
by Hannah Verdin, Barbara D'haene, Diane Beysen, Yana Novikova, Björn Menten, Tom Sante, Pablo Lapunzina, Julian Nevado, Claudia M. B. Carvalho, James R. Lupski, Elfride De Baere
Genomic disorders are often caused by recurrent copy number variations (CNVs), with nonallelic homologous recombination (NAHR) as the underlying mechanism. Recently, several microhomology-mediated repair mechanisms—such as microhomology-mediated end-joining (MMEJ), fork stalling and template switching (FoSTeS), microhomology-mediated break-induced replication (MMBIR), serial replication slippage (SRS), and break-induced SRS (BISRS)—were described in the etiology of non-recurrent CNVs in human disease. In addition, their formation may be stimulated by genomic architectural features. It is, however, largely unexplored to what extent these mechanisms contribute to rare, locus-specific pathogenic CNVs. Here, fine-mapping of 42 microdeletions of the FOXL2 locus, encompassing FOXL2 (32) or its regulatory domain (10), serves as a model for rare, locus-specific CNVs implicated in genetic disease. These deletions lead to blepharophimosis syndrome (BPES), a developmental condition affecting the eyelids and the ovary. For breakpoint mapping we used targeted array-based comparative genomic hybridization (aCGH), quantitative PCR (qPCR), long-range PCR, and Sanger sequencing of the junction products. Microhomology, ranging from 1 bp to 66 bp, was found in 91.7% of 24 characterized breakpoint junctions, being significantly enriched in comparison with a random control sample. Our results show that microhomology-mediated repair mechanisms underlie at least 50% of these microdeletions. Moreover, genomic architectural features, like sequence motifs, non-B DNA conformations, and repetitive elements, were found in all breakpoint regions. In conclusion, the majority of these microdeletions result from microhomology-mediated mechanisms like MMEJ, FoSTeS, MMBIR, SRS, or BISRS. Moreover, we hypothesize that the genomic architecture might drive their formation by increasing the susceptibility for DNA breakage or promote replication fork stalling. Finally, our locus-centered study, elucidating the etiology of a large set of rare microdeletions involved in a monogenic disorder, can serve as a model for other clustered, non-recurrent microdeletions in genetic disease.
RFECS: A Random-Forest Based Algorithm for Enhancer Identification from Chromatin State
RFECS: A Random-Forest Based Algorithm for Enhancer Identification from Chromatin State:
by Nisha Rajagopal, Wei Xie, Yan Li, Uli Wagner, Wei Wang, John Stamatoyannopoulos, Jason Ernst, Manolis Kellis, Bing Ren
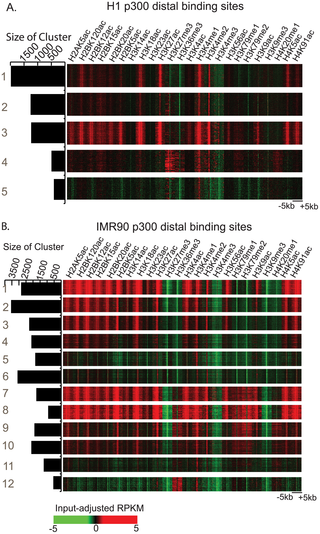
Transcriptional enhancers play critical roles in regulation of gene expression, but their identification in the eukaryotic genome has been challenging. Recently, it was shown that enhancers in the mammalian genome are associated with characteristic histone modification patterns, which have been increasingly exploited for enhancer identification. However, only a limited number of cell types or chromatin marks have previously been investigated for this purpose, leaving the question unanswered whether there exists an optimal set of histone modifications for enhancer prediction in different cell types. Here, we address this issue by exploring genome-wide profiles of 24 histone modifications in two distinct human cell types, embryonic stem cells and lung fibroblasts. We developed a Random-Forest based algorithm, RFECS (Random Forest based Enhancer identification from Chromatin States) to integrate histone modification profiles for identification of enhancers, and used it to identify enhancers in a number of cell-types. We show that RFECS not only leads to more accurate and precise prediction of enhancers than previous methods, but also helps identify the most informative and robust set of three chromatin marks for enhancer prediction.
by Nisha Rajagopal, Wei Xie, Yan Li, Uli Wagner, Wei Wang, John Stamatoyannopoulos, Jason Ernst, Manolis Kellis, Bing Ren
Transcriptional enhancers play critical roles in regulation of gene expression, but their identification in the eukaryotic genome has been challenging. Recently, it was shown that enhancers in the mammalian genome are associated with characteristic histone modification patterns, which have been increasingly exploited for enhancer identification. However, only a limited number of cell types or chromatin marks have previously been investigated for this purpose, leaving the question unanswered whether there exists an optimal set of histone modifications for enhancer prediction in different cell types. Here, we address this issue by exploring genome-wide profiles of 24 histone modifications in two distinct human cell types, embryonic stem cells and lung fibroblasts. We developed a Random-Forest based algorithm, RFECS (Random Forest based Enhancer identification from Chromatin States) to integrate histone modification profiles for identification of enhancers, and used it to identify enhancers in a number of cell-types. We show that RFECS not only leads to more accurate and precise prediction of enhancers than previous methods, but also helps identify the most informative and robust set of three chromatin marks for enhancer prediction.
Nuclear Positioning
Nuclear Positioning: Gregg G. Gundersen, Howard J. Worman. The nucleus is the largest organelle and is commonly depicted in the center of the cell. Yet during cell division, migration, and differentiation, it frequently moves to an asymmetric position ali....
When Lamins Go Bad: Nuclear Structure and Disease
When Lamins Go Bad: Nuclear Structure and Disease: Katherine H. Schreiber, Brian K. Kennedy. Mutations in nuclear lamins or other proteins of the nuclear envelope are the root cause of a group of phenotypically diverse genetic disorders known as laminopathies, which have symptoms that ran....

When Lamins Go Bad: Nuclear Structure and Disease
When Lamins Go Bad: Nuclear Structure and Disease: Katherine H. Schreiber, Brian K. Kennedy. Mutations in nuclear lamins or other proteins of the nuclear envelope are the root cause of a group of phenotypically diverse genetic disorders known as laminopathies, which have symptoms that ran....
Chromatin Movement in the Maintenance of Genome Stability
Chromatin Movement in the Maintenance of Genome Stability: Vincent Dion, Susan M. Gasser. Mechanistic analyses based on improved imaging techniques have begun to explore the biological implications of chromatin movement within the nucleus. Studies in both prokaryotes and eukaryotes hav....
CTCF and Cohesin: Linking Gene Regulatory Elements with Their Targets
CTCF and Cohesin: Linking Gene Regulatory Elements with Their Targets: Matthias Merkenschlager, Duncan T. Odom. Current epigenomics approaches have facilitated the genome-wide identification of regulatory elements based on chromatin features and transcriptional regulator binding and have begun to map long-r....
Transcriptional Regulation and Its Misregulation in Disease
Transcriptional Regulation and Its Misregulation in Disease: Tong Ihn Lee, Richard A. Young. The gene expression programs that establish and maintain specific cell states in humans are controlled by thousands of transcription factors, cofactors, and chromatin regulators. Misregulation of ....

[Report] Adaptive Evolution of Multiple Traits Through Multiple Mutations at a Single Gene
[Report] Adaptive Evolution of Multiple Traits Through Multiple Mutations at a Single Gene: The light color of mice living in the Nebraska Sand Hills is not the result of a single large-effect mutation.
Authors: Catherine R. Linnen, Yu-Ping Poh, Brant K. Peterson, Rowan D. H. Barrett, Joanna G. Larson, Jeffrey D. Jensen, Hopi E. Hoekstra
Authors: Catherine R. Linnen, Yu-Ping Poh, Brant K. Peterson, Rowan D. H. Barrett, Joanna G. Larson, Jeffrey D. Jensen, Hopi E. Hoekstra
Tuesday, March 12, 2013
Structure, evolution and function of the bi-directionally transcribed iab-4/iab-8 microRNA locus in arthropods
Structure, evolution and function of the bi-directionally transcribed iab-4/iab-8 microRNA locus in arthropods:
In Drosophila melanogaster, the iab-4/iab-8 locus encodes bi-directionally transcribed microRNAs that regulate the function of flanking Hox transcription factors. We show that bi-directional transcription, temporal and spatial expression patterns and Hox regulatory function of the iab-4/iab-8 locus are conserved between fly and the beetle Tribolium castaneum. Computational predictions suggest iab-4 and iab-8 microRNAs can target common sites, and cell-culture assays confirm that iab-4 and iab-8 function overlaps on Hox target sites in both fly and beetle. However, we observe key differences in the way Hox genes are targeted. For instance, abd-A transcripts are targeted only by iab-8 in Drosophila, whereas both iab-4 and iab-8 bind to Tribolium abd-A. Our evolutionary and functional characterization of a bi-directionally transcribed microRNA establishes the iab-4/iab-8 system as a model for understanding how multiple products from sense and antisense microRNAs target common sites.
In Drosophila melanogaster, the iab-4/iab-8 locus encodes bi-directionally transcribed microRNAs that regulate the function of flanking Hox transcription factors. We show that bi-directional transcription, temporal and spatial expression patterns and Hox regulatory function of the iab-4/iab-8 locus are conserved between fly and the beetle Tribolium castaneum. Computational predictions suggest iab-4 and iab-8 microRNAs can target common sites, and cell-culture assays confirm that iab-4 and iab-8 function overlaps on Hox target sites in both fly and beetle. However, we observe key differences in the way Hox genes are targeted. For instance, abd-A transcripts are targeted only by iab-8 in Drosophila, whereas both iab-4 and iab-8 bind to Tribolium abd-A. Our evolutionary and functional characterization of a bi-directionally transcribed microRNA establishes the iab-4/iab-8 system as a model for understanding how multiple products from sense and antisense microRNAs target common sites.
Genome-wide localization of exosome components to active promoters and chromatin insulators in Drosophila
Genome-wide localization of exosome components to active promoters and chromatin insulators in Drosophila:
Chromatin insulators are functionally conserved DNA–protein complexes situated throughout the genome that organize independent transcriptional domains. Previous work implicated RNA as an important cofactor in chromatin insulator activity, although the precise mechanisms are not yet understood. Here we identify the exosome, the highly conserved major cellular 3' to 5' RNA degradation machinery, as a physical interactor of CP190-dependent chromatin insulator complexes in Drosophila. Genome-wide profiling of exosome by ChIP-seq in two different embryonic cell lines reveals extensive and specific overlap with the CP190, BEAF-32 and CTCF insulator proteins. Colocalization occurs mainly at promoters but also boundary elements such as Mcp, Fab-8, scs and scs', which overlaps with a promoter. Surprisingly, exosome associates primarily with promoters but not gene bodies of active genes, arguing against simple cotranscriptional recruitment to RNA substrates. Similar to insulator proteins, exosome is also significantly enriched at divergently transcribed promoters. Directed ChIP of exosome in cell lines depleted of insulator proteins shows that CTCF is required specifically for exosome association at Mcp and Fab-8 but not other sites, suggesting that alternate mechanisms must also contribute to exosome chromatin recruitment. Taken together, our results reveal a novel positive relationship between exosome and chromatin insulators throughout the genome.
Chromatin insulators are functionally conserved DNA–protein complexes situated throughout the genome that organize independent transcriptional domains. Previous work implicated RNA as an important cofactor in chromatin insulator activity, although the precise mechanisms are not yet understood. Here we identify the exosome, the highly conserved major cellular 3' to 5' RNA degradation machinery, as a physical interactor of CP190-dependent chromatin insulator complexes in Drosophila. Genome-wide profiling of exosome by ChIP-seq in two different embryonic cell lines reveals extensive and specific overlap with the CP190, BEAF-32 and CTCF insulator proteins. Colocalization occurs mainly at promoters but also boundary elements such as Mcp, Fab-8, scs and scs', which overlaps with a promoter. Surprisingly, exosome associates primarily with promoters but not gene bodies of active genes, arguing against simple cotranscriptional recruitment to RNA substrates. Similar to insulator proteins, exosome is also significantly enriched at divergently transcribed promoters. Directed ChIP of exosome in cell lines depleted of insulator proteins shows that CTCF is required specifically for exosome association at Mcp and Fab-8 but not other sites, suggesting that alternate mechanisms must also contribute to exosome chromatin recruitment. Taken together, our results reveal a novel positive relationship between exosome and chromatin insulators throughout the genome.
Shaping of a morphogenetic gene network [Developmental Biology]
Shaping of a morphogenetic gene network [Developmental Biology]: The Abdominal-B selector protein induces organogenesis of the posterior spiracles by coordinating an organ-specific gene network. The complexity of this network begs the questions of how it originated and what selective pressures drove its formation. Given that the network likely formed in a piecemeal fashion, with elements recruited sequentially, we...
Temporal control of BMP signalling determines neuronal subtype identity in the dorsal neural tube [RESEARCH ARTICLES]
Temporal control of BMP signalling determines neuronal subtype identity in the dorsal neural tube [RESEARCH ARTICLES]: Samuel Tozer, Gwenvael Le Dreau, Elisa Marti, and James Briscoe
The conventional explanation for how a morphogen patterns a tissue holds that cells interpret different concentrations of an extrinsic ligand by producing corresponding levels of intracellular signalling activity, which in turn regulate differential gene expression. However, this view has been challenged, raising the possibility that distinct mechanisms are used to interpret different morphogens. Here, we investigate graded BMP signalling in the vertebrate neural tube. We show that defined exposure times to Bmp4 generate distinct levels of signalling and induce specific dorsal identities. Moreover, we provide evidence that a dynamic gradient of BMP activity confers progressively more dorsal neural identities in vivo. These results highlight a strategy for morphogen interpretation in which the tight temporal control of signalling is important for the spatial pattern of cellular differentiation.
The conventional explanation for how a morphogen patterns a tissue holds that cells interpret different concentrations of an extrinsic ligand by producing corresponding levels of intracellular signalling activity, which in turn regulate differential gene expression. However, this view has been challenged, raising the possibility that distinct mechanisms are used to interpret different morphogens. Here, we investigate graded BMP signalling in the vertebrate neural tube. We show that defined exposure times to Bmp4 generate distinct levels of signalling and induce specific dorsal identities. Moreover, we provide evidence that a dynamic gradient of BMP activity confers progressively more dorsal neural identities in vivo. These results highlight a strategy for morphogen interpretation in which the tight temporal control of signalling is important for the spatial pattern of cellular differentiation.
Conserved non-coding elements and cis regulation: actions speak louder than words [REVIEWS]
Conserved non-coding elements and cis regulation: actions speak louder than words [REVIEWS]: Andrew C. Nelson and Fiona C. Wardle
It is a truth (almost) universally acknowledged that conserved non-coding genomic sequences function in the cis regulation of neighbouring genes. But is this a misconception? The literature is strewn with examples of conserved non-coding sequences being able to drive reporter expression, but the extent to which such sequences are actually used endogenously in vivo is only now being rigorously explored using unbiased genome-scale approaches. Here, we review the emerging picture, examining the extent to which conserved non-coding sequences equivalently regulate gene expression in different species, or at different developmental stages, and how genomics approaches are revealing the relationship between sequence conservation and functional use of cis-regulatory elements.
It is a truth (almost) universally acknowledged that conserved non-coding genomic sequences function in the cis regulation of neighbouring genes. But is this a misconception? The literature is strewn with examples of conserved non-coding sequences being able to drive reporter expression, but the extent to which such sequences are actually used endogenously in vivo is only now being rigorously explored using unbiased genome-scale approaches. Here, we review the emerging picture, examining the extent to which conserved non-coding sequences equivalently regulate gene expression in different species, or at different developmental stages, and how genomics approaches are revealing the relationship between sequence conservation and functional use of cis-regulatory elements.
Friday, March 1, 2013
A chromatin link to caste identity in the carpenter ant Camponotus floridanus [RESEARCH]
A chromatin link to caste identity in the carpenter ant Camponotus floridanus [RESEARCH]:
In many ant species, sibling larvae follow alternative ontogenetic trajectories that generate striking variation in morphology and behavior among adults. These organism-level outcomes are often determined by environmental rather than genetic factors. Therefore, epigenetic mechanisms may mediate the expression of adult polyphenisms. We produced the first genome-wide maps of chromatin structure in a eusocial insect and found that gene-proximal changes in histone modifications, notably H3K27 acetylation, discriminate two female worker and male castes in Camponotus floridanus ants and partially explain differential gene expression between castes. Genes showing coordinated changes in H3K27ac and RNA implicate muscle development, neuronal regulation, and sensory responses in modulating caste identity. Binding sites of the acetyltransferase CBP harbor the greatest caste variation in H3K27ac, are enriched with motifs for conserved transcription factors, and show evolutionary expansion near developmental and neuronal genes. These results suggest that environmental effects on caste identity may be mediated by differential recruitment of CBP to chromatin. We propose that epigenetic mechanisms that modify chromatin structure may help orchestrate the generation and maintenance of polyphenic caste morphology and social behavior in ants.

In many ant species, sibling larvae follow alternative ontogenetic trajectories that generate striking variation in morphology and behavior among adults. These organism-level outcomes are often determined by environmental rather than genetic factors. Therefore, epigenetic mechanisms may mediate the expression of adult polyphenisms. We produced the first genome-wide maps of chromatin structure in a eusocial insect and found that gene-proximal changes in histone modifications, notably H3K27 acetylation, discriminate two female worker and male castes in Camponotus floridanus ants and partially explain differential gene expression between castes. Genes showing coordinated changes in H3K27ac and RNA implicate muscle development, neuronal regulation, and sensory responses in modulating caste identity. Binding sites of the acetyltransferase CBP harbor the greatest caste variation in H3K27ac, are enriched with motifs for conserved transcription factors, and show evolutionary expansion near developmental and neuronal genes. These results suggest that environmental effects on caste identity may be mediated by differential recruitment of CBP to chromatin. We propose that epigenetic mechanisms that modify chromatin structure may help orchestrate the generation and maintenance of polyphenic caste morphology and social behavior in ants.
An Integrated Holo-Enhancer Unit Defines Tissue and Gene Specificity of the Fgf8 Regulatory Landscape
An Integrated Holo-Enhancer Unit Defines Tissue and Gene Specificity of the Fgf8 Regulatory Landscape: Mirna Marinić, Tugce Aktas, Sandra Ruf, François Spitz. Fgf8 encodes a key signaling factor, and its precise regulation is essential for embryo patterning. Here, we identified the regulatory modules that control Fgf8 expression during mam....
H3K4me3 Interactions with TAF3 Regulate Preinitiation Complex Assembly and Selective Gene Activation
H3K4me3 Interactions with TAF3 Regulate Preinitiation Complex Assembly and Selective Gene Activation: Shannon M. Lauberth, Takahiro Nakayama, Xiaolin Wu, Andrea L. Ferris, Zhanyun Tang, Stephen H. Hughes, Robert G. Roeder. Histone modifications regulate chromatin-dependent processes, yet the mechanisms by which they contribute to specific outcomes remain unclear. H3K4me3 is a prominent histone mark that is associate....

Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression
Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression: Lei S. Qi, Matthew H. Larson, Luke A. Gilbert, Jennifer A. Doudna, Jonathan S. Weissman, Adam P. Arkin, Wendell A. Lim. Targeted gene regulation on a genome-wide scale is a powerful strategy for interrogating, perturbing, and engineering cellular systems. Here, we develop a method for controlling gene expression ba....
Subscribe to:
Comments (Atom)