Justin Crocker's reading list...
Wednesday, January 4, 2017
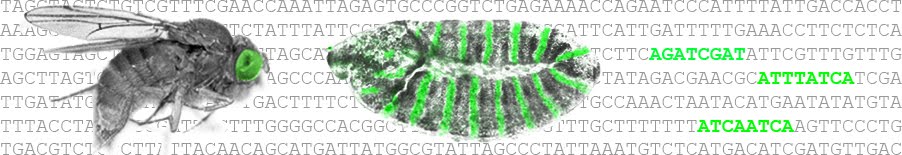
A Fully Synthetic Transcriptional Platform for a Multicellular Eukaryote
Monday, August 12, 2013
Moving Reading List
For those of you who follow this list, I'll be moving it over to twitter--it's far easier to manage: https://twitter.com/JustinMCrocker
Sunday, August 4, 2013
The landscape of RNA polymerase II transcription initiation in C. elegansreveals promoter and enhancer architectures
The landscape of RNA polymerase II transcription initiation in C. elegansreveals promoter and enhancer architectures

RNA polymerase transcription initiation sites are largely unknown inCaenorhabditis elegans. The initial 5′ end of most protein-coding transcripts is removed by trans-splicing, and noncoding initiation sites have not been investigated. We characterized the landscape of RNA Pol II transcription initiation, identifying 73,500 distinct clusters of initiation. Bidirectional transcription is frequent, with a peak of transcriptional pairing at 120 bp. We assign transcription initiation sites to 7691 protein-coding genes and find that they display features typical of eukaryotic promoters. Strikingly, the majority of initiation events occur in regions with enhancer-like chromatin signatures. Based on the overlap of transcription initiation clusters with mapped transcription factor binding sites, we define 2361 transcribed intergenic enhancers. Remarkably, productive transcription elongation across these enhancers is predominantly in the same orientation as that of the nearest downstream gene. Directed elongation from an upstream enhancer toward a downstream gene could potentially deliver RNA polymerase II to a proximal promoter, or alternatively might function directly as a distal promoter. Our results provide a new resource to investigate transcription regulation in metazoans.
Social insect genomes exhibit dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality
Genomes of eusocial insects code for dramatic examples of phenotypic plasticity and social organization. We compared the genomes of seven ants, the honeybee, and various solitary insects to examine whether eusocial lineages share distinct features of genomic organization. Each ant lineage contains ~4000 novel genes, but only 64 of these genes are conserved among all seven ants. Many gene families have been expanded in ants, notably those involved in chemical communication (e.g., desaturases and odorant receptors). Alignment of the ant genomes revealed reduced purifying selection compared withDrosophila without significantly reduced synteny. Correspondingly, ant genomes exhibit dramatic divergence of noncoding regulatory elements; however, extant conserved regions are enriched for novel noncoding RNAs and transcription factor–binding sites. Comparison of orthologous gene promoters between eusocial and solitary species revealed significant regulatory evolution in both cis (e.g., Creb) and trans (e.g., fork head) for nearly 2000 genes, many of which exhibit phenotypic plasticity. Our results emphasize that genomic changes can occur remarkably fast in ants, because two recently diverged leaf-cutter ant species exhibit faster accumulation of species-specific genes and greater divergence in regulatory elements compared with other ants or Drosophila. Thus, while the "socio-genomes" of ants and the honeybee are broadly characterized by a pervasive pattern of divergence in gene composition and regulation, they preserve lineage-specific regulatory features linked to eusociality. We propose that changes in gene regulation played a key role in the origins of insect eusociality, whereas changes in gene composition were more relevant for lineage-specific eusocial adaptations.
Genomes of eusocial insects code for dramatic examples of phenotypic plasticity and social organization. We compared the genomes of seven ants, the honeybee, and various solitary insects to examine whether eusocial lineages share distinct features of genomic organization. Each ant lineage contains ~4000 novel genes, but only 64 of these genes are conserved among all seven ants. Many gene families have been expanded in ants, notably those involved in chemical communication (e.g., desaturases and odorant receptors). Alignment of the ant genomes revealed reduced purifying selection compared withDrosophila without significantly reduced synteny. Correspondingly, ant genomes exhibit dramatic divergence of noncoding regulatory elements; however, extant conserved regions are enriched for novel noncoding RNAs and transcription factor–binding sites. Comparison of orthologous gene promoters between eusocial and solitary species revealed significant regulatory evolution in both cis (e.g., Creb) and trans (e.g., fork head) for nearly 2000 genes, many of which exhibit phenotypic plasticity. Our results emphasize that genomic changes can occur remarkably fast in ants, because two recently diverged leaf-cutter ant species exhibit faster accumulation of species-specific genes and greater divergence in regulatory elements compared with other ants or Drosophila. Thus, while the "socio-genomes" of ants and the honeybee are broadly characterized by a pervasive pattern of divergence in gene composition and regulation, they preserve lineage-specific regulatory features linked to eusociality. We propose that changes in gene regulation played a key role in the origins of insect eusociality, whereas changes in gene composition were more relevant for lineage-specific eusocial adaptations.
Inferring chromatin-bound protein complexes from genome-wide binding assays
|
|

Genome-wide binding assays can determine where individual transcription factors bind in the genome. However, these factors rarely bind chromatin alone, but instead frequently bind to cis-regulatory elements (CREs) together with other factors thus forming protein complexes. Currently there are no integrative analytical approaches that can predict which complexes are formed on chromatin. Here, we describe a computational methodology to systematically capture protein complexes and infer their impact on gene expression. We applied our method to three human cell types, identified thousands of CREs, inferred known and undescribed complexes recruited to these CREs, and determined the role of the complexes as activators or repressors. Importantly, we found that the predicted complexes have a higher number of physical interactions between their members than expected by chance. Our work provides a mechanism for developing hypotheses about gene regulation via binding partners, and deciphering the interplay between combinatorial binding and gene expression.
Monday, July 29, 2013
Friday, July 26, 2013
Local features determine cis-regulatory function [Genetics]
by White, M. A., Myers, C. A., Corbo, J. C., Cohen, B. A.
Subscribe to:
Posts (Atom)